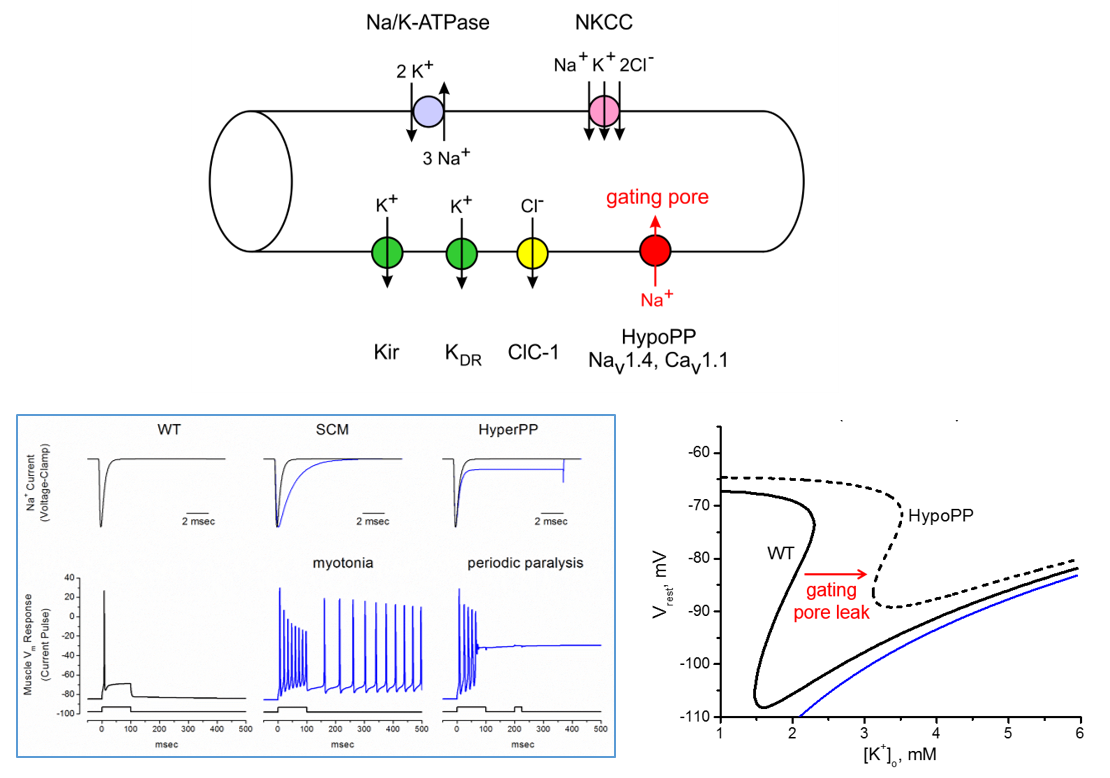
A major effort in our lab is to define the functional consequences of ion channel mutations identified in patients with myotonia or periodic paralysis. Functional testing is performed by expressing mutant ion channels is cells (HEK) or frog eggs (Xenopus oocytes), and then recording ionic currents under voltage-clamp control of the cell. With this approach we have established:
Sodium channel (NaV1.4) gain of function defects in autosomal dominant disorders:
• Myotonia mutations typically cause:
o Impaired inactivation (onset too slow, recovery too fast, or mild depolarized shift in voltage dependence).
o Mildly enhanced activation (very small hyperpolarized shift in voltage dependence
• Hyperkalemic periodic paralysis / Paramyotonia congenita mutations cause:
o Impaired inactivation (onset too slow, recovery too fast, depolarized shift in voltage dependence, failure to completely inactivate ~1% to 4% persistent Na current)
o Impaired slow inactivation (less complete, depolarized voltage dependence)
Sodium channel (NaV1.4) loss of function defects in autosomal recessive disorders:
• Congenital myasthenic syndrome are associated with:
o Enhanced inactivation (hyperpolarized shift in voltage dependence, slower recovery from inactivation)
• Congenital myopathy with hypotonia and hypokinesia are associated with (F. Mutoni group):
o Severe loss of function defects (null mutation or loss of function with decreased current density)
“Leaky” mutant channels caused by defects in the voltage-sensor domains of the skeletal muscle calcium channel (CaV1.1) and sodium channel (NaV1.4) in Hypokalemic periodic paralysis.